
This blog post is intended for geneticists and dataflow engineers who need to compare genetic variants.
Have you ever tried to determine if two genetic variants are the same? If so, you’re not alone. There are competing ways to represent variants, handling ambiguous assignments, as well as reconciling updates to underlying sequence models. To help you with these problems, we’re introducing a new set of web services for comparing and grouping variants.
In this post we’ll briefly describe the services and run through a simple example. For those interested in more detail, we’ve added a description of our new data format, SPDI, at the end. (SPDI stands for Sequence-ID, Position, Deleted sequence, Inserted sequence. You’ll see why below.)
There’s also full documentation available if you’d rather jump right in.
What can the services do?
The services are grouped into three sets based on the type of input file they require:
- SPDI (our new data model—see below)
- HGVS
- VCF
Most of the services work with SPDI. The services for HGVS and VCF basically convert your data into SPDI format for use in the other services.
The SPDI services can do the following:
- Produce a contextual allele, a new feature of our SPDI format that corrects left/right-shifting or shuffling for insertions or deletions
- Transform data into right-shifted HGVS
- Transform data into left-shifted VCF fields (seq-id, 1-based position, reference and alt alleles, with appropriate anchor nucleotides)
- Remap (or lift-over) a variant to all available locations based on the alignment dataset used by ClinVar and dbSNP
- Remap (or lift-over) to a canonical representative at a single location that you can use to group identical variants
Let’s see an example!
Suppose you have this VCF data for a variant:
#Assm=GCF_000001405.25 #CHROM POS ID REF ALT QUAL FILTER INFO 11 5248224 id1 A AC . . .
Suppose you also have an HGVS expression for another variant:
NM_000518.4:c.27dupG
Are these two the same variant?
The VCF indicates that a C is inserted on chromosome 11 of GRCh37p13 (hg19).
The HGVS indicates that a G is inserted on NM_000518.4, the transcript for the beta subunit of human hemoglobin (HBB).
Let’s first use the VCF and HGVS services to convert both expressions to SPDI.
(base URL: https://api.ncbi.nlm.nih.gov/variation/v0/)
GET hgvs/NM_000518.4:c.27dupG/contextuals | |-> NM_000518.4:76:G:GG GET vcf/11/5248224/A/AC/contextuals?assembly=GCF_000001405.25 | |-> NC_000011.9:5248224:C:CC
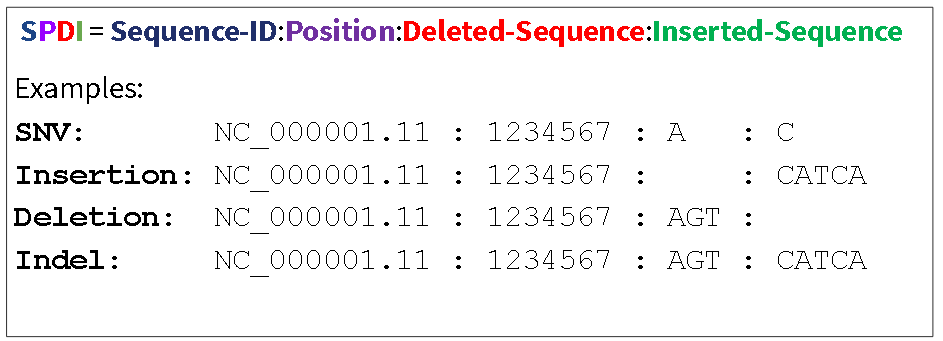
SPDI contains four fields separated by a colon (:): sequence id, position, deleted sequence, and inserted sequence.
To find out if these variants are the same, we input the SPDI to find the canonical representative for each variant:
GET spdi/NM_000518.4:76:G:GG/canonical_representative | |-> NC_000011.10:5226994:C:CC GET spdi/NC_000011.9:5248224:C:CC/canonical_representative | |-> NC_000011.10:5226994:C:CC
The canonical representatives are the same!
Try these new services out and let us know what you think. You can comment on this post, or write to us.
FOR MORE INFORMATION
Glossary
- Allele: A particular biological sequence at a particular interval within a larger sequence.
- Canonical Representative: A representation of a variant using a defined sequence model. When remapped to the same model, identical variants will have the same canonical representative.
- Contextual Allele: An allele that is corrected for over-precision.
- SPDI: Sequence-id, Position, Deleted Sequence, Insertion Sequence is a simple data serialization format for variants with two known breakpoints (end-points).
Appendix: The SPDI data model
The SPDI format encodes variants using four fields (Figure 1). Variants are represented as the deletion of a sequence interval and a replacement with an inserted sequence. Either the deleted or inserted interval can be empty, producing a pure insertion or deletion. The position is a 0-based coordinate for where the deletion starts. This means that position 0 indicates a deletion that starts immediately before the first nucleotide, and position 1 a deletion that occurs between residues 1 and 2, and so on.
These services return SPDI data as a structure:
{
“seq_id”: “NM_000518.4”,
“position”: 76,
“deletion-sequence”: “G”,
“insertion-sequence”: “GG”
}
The central function of these new services is producing a contextual allele. Such alleles correct any over-precision (left/right-shifting or shuffling) that can occur in descriptions of pure insertions and deletions (Figure 2). Please note that simply because a variant is represented as SPDI does not guarantee that it represents a contextual allele. Therefore, any SPDI expression provided to these services is first converted to a contextual allele before further processing.

2 thoughts on “New Web Services for Comparing and Grouping Sequence Variants”